
A fibrilhação auricular (FA) desenvolve-se em aurículas estrutural e eletricamente alteradas.
| Principais Mecanismos da Fibrilhação Auricular em Síndromes Arrítmicas Hereditárias e Cardiomiopatias | ||
|---|---|---|
| Diagnósticos | Alterações auriculares | Mecanismo de desenvolvimento da FA |
| CMH CMD CAVD |
Alterações estruturais auriculares (dilatação, fibrose, aumento do stress parietal) |
O remodelamento estrutural cria um substrato arritmogénico para arritmias auriculares e fibrilhação auricular. |
|
Síndrome do QT longo Síndrome do QT curto Síndrome de Brugada TVPC Síndrome de WPW |
Alterações elétricas e iónicas nas aurículas | A disfunção dos canais iónicos e a instabilidade elétrica aumentam a vulnerabilidade auricular a arritmias e FA. |
|
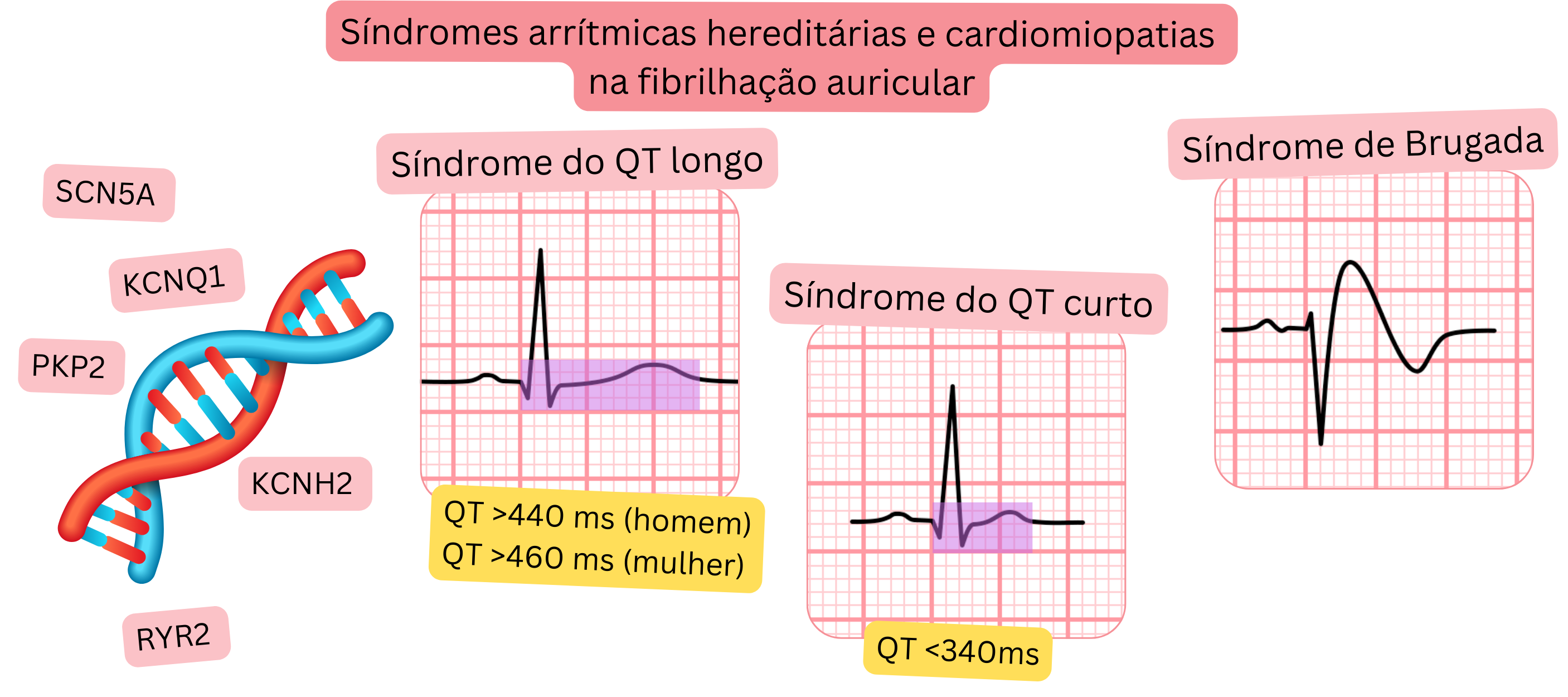
Mutações genéticas (SCN5A, KCNQ1, KCNH2, RYR2, etc.) |
Alterações genéticas que afetam o miocárdio auricular | As mutações envolvem não só os ventrículos, mas também as aurículas, criando assim um substrato arritmogénico para FA. |
| CMH CMD |
Sobrecarga crónica e disfunção diastólica | O aumento das pressões de enchimento conduz à dilatação da aurícula esquerda e à formação de um substrato estável para FA. |
| TVPC Síndrome do QT longo |
Disregulação autonómica | O stress adrenérgico e a regulação autonómica comprometida promovem arritmias auriculares e FA. |
CAVD – Cardiomiopatia arritmogénica do ventrículo direito, TVPC – Taquicardia ventricular polimórfica catecolaminérgica, CMD – Cardiomiopatia dilatada, FA – Fibrilhação auricular, CMH – Cardiomiopatia hipertrófica
As síndromes arrítmicas e as cardiomiopatias também podem surgir devido a mutações genéticas.
| Mutações Genéticas e a Sua Associação com Fibrilhação Auricular | |||
|---|---|---|---|
| Mutação genética | Prevalência de FA | Diagnósticos cardíacos associados | Mecanismo da FA |
| SCN5A | 20–40 % |
Síndrome de Brugada CMD (fenótipo elétrico) FA genética |
Instabilidade elétrica auricular. |
| KCNQ1 | 10–30 % |
Síndrome do QT longo tipo 1 FA familiar |
Anomalia da repolarização auricular. |
| KCNH2 (HERG) | 10–25 % | Síndrome do QT longo tipo 2 | Aumento da vulnerabilidade auricular. |
| RYR2 | 5–15 % | TVPC | Descargas adrenérgicas de Ca2+ → ectopia. |
| CACNA1C | 30–50 % |
Fenótipo de Brugada Síndrome de Timothy Arritmias auriculares |
Disfunção do canal de Ca2+ → excitabilidade auricular. |
| MYH7 | 20–35 % |
CMH CMD CMVNE |
Fibrose e dilatação auriculares. |
| MYBPC3 | 20–35 % | CMH | Fibrose auricular na CMH. |
| LMNA | 45–70 % | Laminopatia (CMD + bloqueio AV) | Remodelamento auricular. |
| TNNI3 | 15–30 % | CMH | Remodelamento auricular na hipertrofia. |
| TNNT2 | 20–35 % |
CMH CMD |
Fibrose auricular. |
| PLN | 15–25 % |
CMD Fenótipo tipo CAVD |
Instabilidade elétrica auricular. |
| DSP | 10–25 % | CAVD | Remodelamento fibrótico auricular. |
| PKP2 | 10–20 % | CAVD | Arritmias auriculares durante a progressão da doença. |
| PRKAG2 | 20–40 % |
Cardiomiopatia de armazenamento de glicogénio Hipertrofia + fenótipo WPW |
Hipertrofia auricular e pré-excitação. |
| GLA | 30–60 % | Doença de Fabry | Fibrose auricular. |
CAVD – Cardiomiopatia arritmogénica do ventrículo direito, TVPC – Taquicardia ventricular polimórfica catecolaminérgica, CMD – Cardiomiopatia dilatada, FA – Fibrilhação auricular, CMH – Cardiomiopatia hipertrófica, CMVNE – Cardiomiopatia por não compactação do ventrículo esquerdo, WPW – Síndrome de Wolff–Parkinson–White
Na tabela seguinte, pode rever a prevalência de FA em síndromes arrítmicas e cardiomiopatias.
| Síndromes Arritmológicas Genéticas e Hereditárias – Prevalência, Risco de FA, Contraindicações e Anticoagulação | ||||
|---|---|---|---|---|
| Diagnóstico | Prevalência | Prevalência de FA | Contraindicações | Anticoagulação |
| Síndrome do QT longo | 1 : 2 000 | 2–29 % | Fármacos que prolongam o QT (1) | De acordo com CHA2DS2-VA |
| Síndrome do QT curto | 1 : 100 000 | 18–70 % | – | De acordo com CHA2DS2-VA |
| Síndrome de Brugada | 1 : 5 000 | 6–53 % | Fármacos antiarrítmicos de classe IC | De acordo com CHA2DS2-VA |
| TVPC | 1 : 50 000 | 11–37 % | – | De acordo com CHA2DS2-VA |
| CMH | 1 : 500 | 17–30 % | Fármacos antiarrítmicos de classe IC | Sempre (NOAC ou varfarina) |
| CAVD | 1 : 2 000 | 9–30 % | – | De acordo com CHA2DS2-VA |
| CMD (mutação LMNA) | 1 : 400 | 25–49 % | – | De acordo com CHA2DS2-VA |
| Síndrome de WPW | 1 : 500 | 7–50 % | Fármacos bloqueadores do nó AV (2) | De acordo com CHA2DS2-VA |
TVPC – Taquicardia ventricular polimórfica catecolaminérgica, CMH – Cardiomiopatia hipertrófica, CAVD – Cardiomiopatia arritmogénica do ventrículo direito, CMD – Cardiomiopatia dilatada.
1 Na síndrome do QT longo, os fármacos que prolongam o QT são contraindicados:
- IA: Quinidina, Procainamida, Disopiramida
- III: Amiodarona, Sotalol, Dofetilida, Ibutilida, Dronedarona
- IC: Propafenona, Flecainida (contraindicação relativa)
2 Fármacos bloqueadores do nó AV: Betabloqueadores, Digoxina, Verapamil, Diltiazem, Amiodarona, Adenosina
Estas diretrizes são não oficiais e não representam diretrizes formais emitidas por qualquer sociedade profissional de cardiologia. Destinam-se apenas a fins educacionais e informativos.
